treeArches: learning and updating a cell-type hierarchy (basic tutorial)
In this tutorial, we explain the different functionalities of treeArches. We show how to:
Step 1: Integrate reference datasets using scVI
Step 2: Match the cell-types in the reference datasets to learn the cell-type hierarchy of the reference datasets using scHPL
Step 3: Apply architural surgery to extend the reference dataset using scArches
Step 4a: Update the learned hierarchy with the cell-types from the query dataset using scHPL (useful when the query dataset is labeled)
Step 4b: Predict the labels of the cells in the query dataset using scHPL (useful when the query dataset is unlabeled)
[2]:
import os
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
warnings.simplefilter(action='ignore', category=UserWarning)
[3]:
import scanpy as sc
import torch
import scarches as sca
from scarches.dataset.trvae.data_handling import remove_sparsity
import matplotlib.pyplot as plt
import numpy as np
import gdown
import copy as cp
import seaborn as sns
WARNING:root:In order to use the mouse gastrulation seqFISH datsets, please install squidpy (see https://github.com/scverse/squidpy).
WARNING:root:In order to use sagenet models, please install pytorch geometric (see https://pytorch-geometric.readthedocs.io) and
captum (see https://github.com/pytorch/captum).
WARNING:root:mvTCR is not installed. To use mvTCR models, please install it first using "pip install mvtcr"
WARNING:root:multigrate is not installed. To use multigrate models, please install it first using "pip install multigrate".
[4]:
sc.settings.set_figure_params(dpi=1000, frameon=False)
sc.set_figure_params(dpi=1000)
sc.set_figure_params(figsize=(7,7))
torch.set_printoptions(precision=3, sci_mode=False, edgeitems=7)
import matplotlib
matplotlib.rcParams['pdf.fonttype'] = 42
Download raw Dataset
[5]:
url = 'https://drive.google.com/uc?id=1LaYOadbotGC6gXAlo-aKfHz-spoFnawk'
output = 'pbmc.h5ad'
gdown.download(url, output, quiet=False)
Downloading...
From: https://drive.google.com/uc?id=1LaYOadbotGC6gXAlo-aKfHz-spoFnawk
To: /Users/chelseaalexandra.bright/theislab/scarches/notebooks/pbmc.h5ad
100%|██████████| 2.06G/2.06G [00:41<00:00, 49.6MB/s]
[5]:
'pbmc.h5ad'
[5]:
adata = sc.read('pbmc.h5ad')
[6]:
adata.X = adata.layers["counts"].copy()
[7]:
adata = adata[adata.obs.study != "Villani"]
We now split the data into reference and query dataset to simulate the building process. Here we use the ‘10X’ batch as query data.
[8]:
target_conditions = ["10X"]
source_adata = adata[~adata.obs.study.isin(target_conditions)].copy()
target_adata = adata[adata.obs.study.isin(target_conditions)].copy()
print(source_adata)
print(target_adata)
AnnData object with n_obs × n_vars = 21757 × 12303
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue'
layers: 'counts'
AnnData object with n_obs × n_vars = 10727 × 12303
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue'
layers: 'counts'
For a better model performance it is necessary to select HVGs. We are doing this by applying the function scanpy.pp.highly_variable_genes(). The parameter n_top_genes is set to 2000 here. However, for more complicated datasets you might have to increase number of genes to capture more diversity in the data.
[9]:
source_adata.raw = source_adata
[10]:
source_adata
[10]:
AnnData object with n_obs × n_vars = 21757 × 12303
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue'
layers: 'counts'
[11]:
sc.pp.normalize_total(source_adata)
[12]:
sc.pp.log1p(source_adata)
[13]:
sc.pp.highly_variable_genes(
source_adata,
n_top_genes=2000,
batch_key="study",
subset=True)
For consistency we set adata.X to be raw counts. In other datasets that may be already the case
[14]:
source_adata.X = source_adata.raw[:, source_adata.var_names].X
Create scVI model and train it on reference dataset
Remember: The adata object has to have count data in adata.X for scVI/scANVI if not further specified.
[15]:
sca.models.SCVI.setup_anndata(source_adata, batch_key="batch")
The scVI model uses the zero-inflated negative binomial (ZINB) loss by default. Insert gene_likelihood='nb' to change the reconstruction loss to negative binomial (NB) loss.
[16]:
vae = sca.models.SCVI(
source_adata,
n_layers=2,
encode_covariates=True,
deeply_inject_covariates=True,
use_layer_norm="both",
use_batch_norm="none",
)
[17]:
vae.train(max_epochs=80)
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 80/80: 100%|██████████████████| 80/80 [01:34<00:00, 1.18s/it, loss=565, v_num=1]
The resulting latent representation of the data can then be visualized with UMAP
[18]:
reference_latent = sc.AnnData(vae.get_latent_representation())
reference_latent.obs["cell_type"] = source_adata.obs["final_annotation"].tolist()
reference_latent.obs["batch"] = source_adata.obs["batch"].tolist()
reference_latent.obs["study"] = source_adata.obs["study"].tolist()
[19]:
sc.pp.neighbors(reference_latent, n_neighbors=8)
sc.tl.leiden(reference_latent)
sc.tl.umap(reference_latent)
[20]:
reference_latent.obs['study'] = reference_latent.obs['study'].astype('category')
# Reorder categories, so smallest dataset is plotted on top
reference_latent.obs['study'].cat.reorder_categories(['Oetjen', 'Sun', 'Freytag'], inplace=True)
[21]:
sc.pl.umap(reference_latent,
color=['study'],
frameon=False,
wspace=0.6, s=25,
palette=sns.color_palette('colorblind', as_cmap=True)[:3]
)
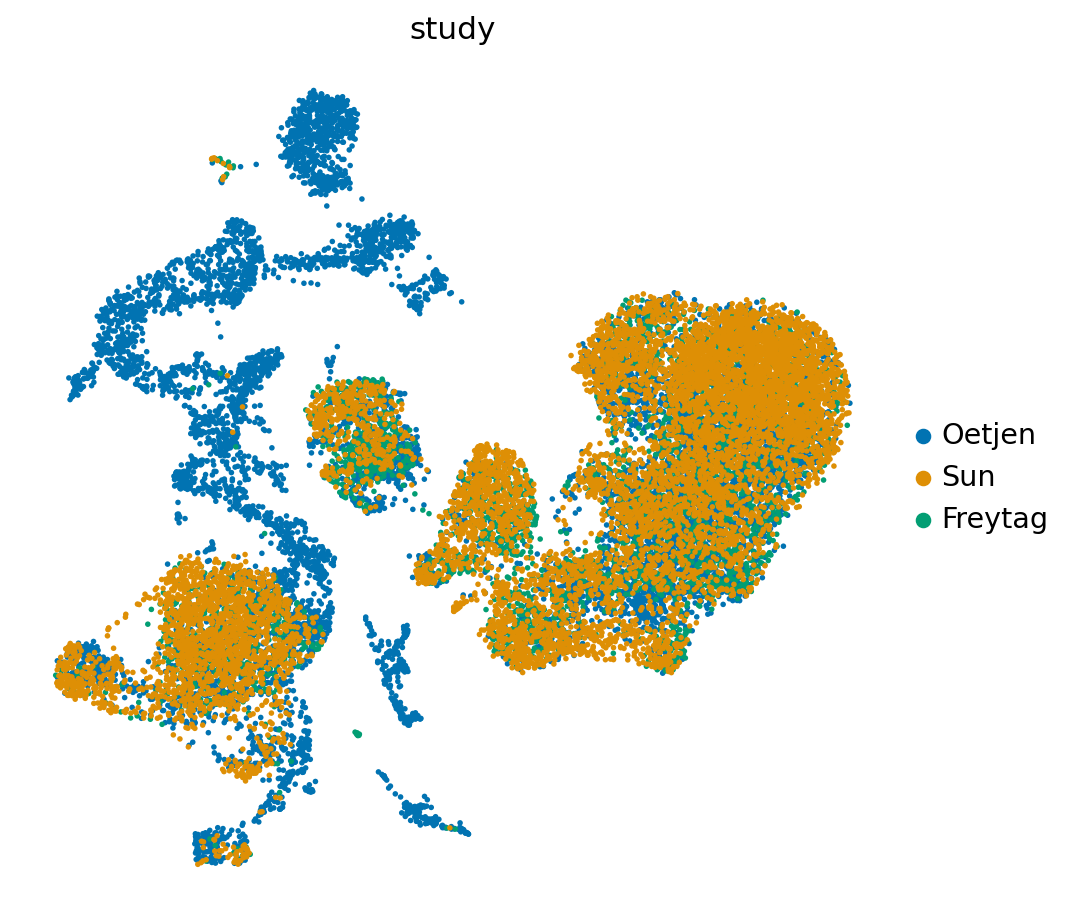
The colorblind color map only contains 10 different colors. To visualize the different cell-types, we rename some cells to a lower resolution.
[22]:
reference_latent.obs['ct_low'] = 0
idx = ((reference_latent.obs.cell_type == 'CD4+ T cells') |
(reference_latent.obs.cell_type == 'CD8+ T cells'))
reference_latent.obs['ct_low'][idx] = 'T cells'
idx = ((reference_latent.obs.cell_type == 'CD10+ B cells') |
(reference_latent.obs.cell_type == 'CD20+ B cells'))
reference_latent.obs['ct_low'][idx] = 'B cells'
idx = ((reference_latent.obs.cell_type == 'CD14+ Monocytes') |
(reference_latent.obs.cell_type == 'CD16+ Monocytes') |
(reference_latent.obs.cell_type == 'Monocyte progenitors'))
reference_latent.obs['ct_low'][idx] = 'Monocytes'
idx = ((reference_latent.obs.cell_type == 'Erythrocytes') |
(reference_latent.obs.cell_type == 'Erythroid progenitors'))
reference_latent.obs['ct_low'][idx] = 'Erythrocytes'
idx = ((reference_latent.obs.cell_type == 'Monocyte-derived dendritic cells') |
(reference_latent.obs.cell_type == 'Plasmacytoid dendritic cells'))
reference_latent.obs['ct_low'][idx] = 'Dendritic cells'
idx = reference_latent.obs.cell_type == 'HSPCs'
reference_latent.obs['ct_low'][idx] = 'HSPCs'
idx = reference_latent.obs.cell_type == 'Megakaryocyte progenitors'
reference_latent.obs['ct_low'][idx] = 'Megakaryocyte progenitors'
idx = reference_latent.obs.cell_type == 'NK cells'
reference_latent.obs['ct_low'][idx] = 'NK cells'
idx = reference_latent.obs.cell_type == 'NKT cells'
reference_latent.obs['ct_low'][idx] = 'NKT cells'
idx = reference_latent.obs.cell_type == 'Plasma cells'
reference_latent.obs['ct_low'][idx] = 'Plasma cells'
/tmp/ipykernel_1063462/4260835584.py:5: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
reference_latent.obs['ct_low'][idx] = 'T cells'
[23]:
sc.pl.umap(reference_latent,
color=['ct_low'],
frameon=False,
wspace=0.6, s=60,
palette=sns.color_palette('colorblind', as_cmap=True)
)

We can also visualize the cell-types per dataset.
[24]:
for s in np.unique(reference_latent.obs.study):
ref_s = cp.deepcopy(reference_latent)
ref_s.obs.ct_low[reference_latent.obs.study != s] = np.nan
sc.pl.umap(ref_s,
color=['ct_low'],
frameon=False,
wspace=0.6, s=60,
palette=sns.color_palette('colorblind', as_cmap=True), title=s,
save=s+'.pdf'
)
WARNING: saving figure to file figures/umapFreytag.pdf
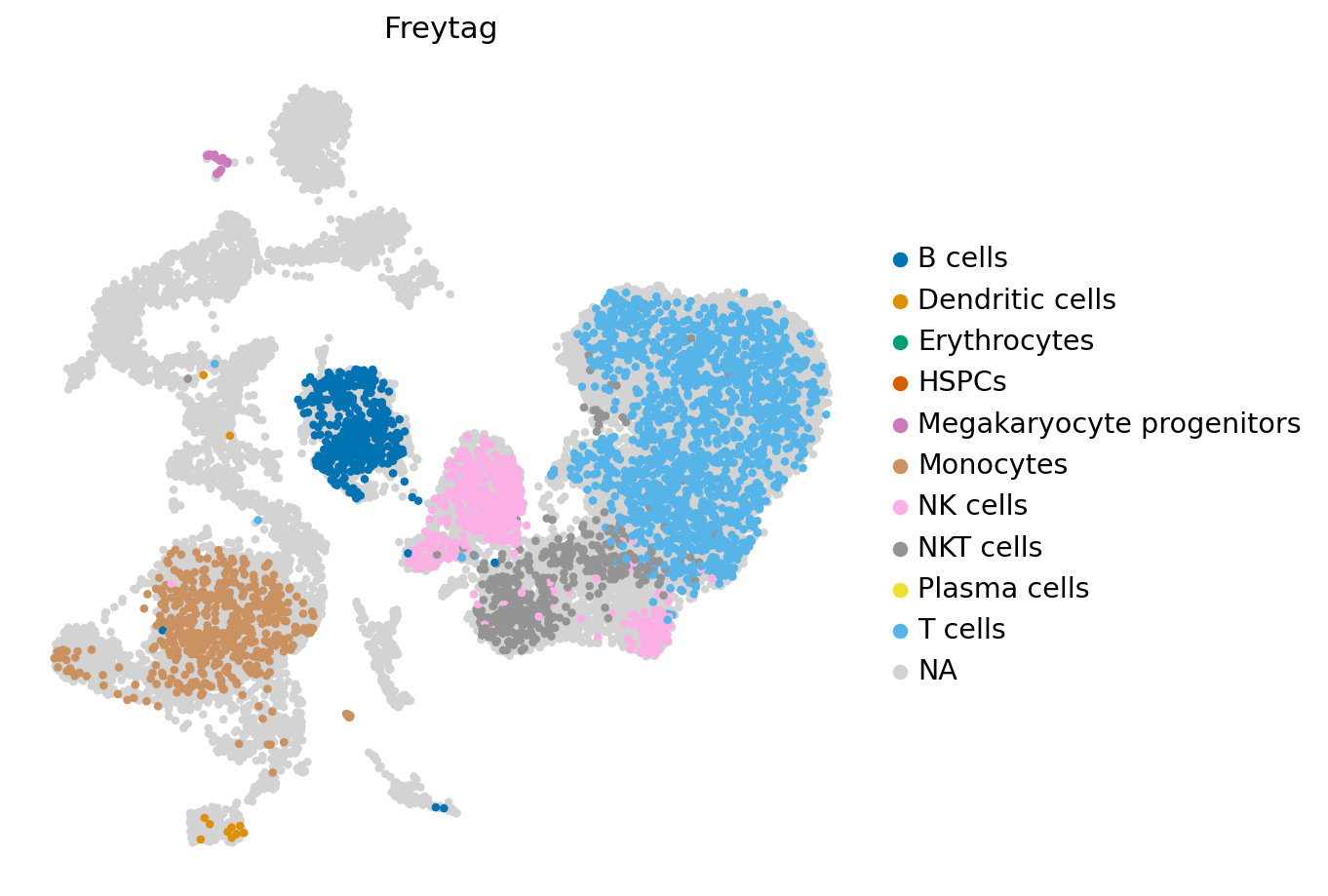
WARNING: saving figure to file figures/umapOetjen.pdf

WARNING: saving figure to file figures/umapSun.pdf

After pretraining the model can be saved for later use or also be uploaded for other researchers with via Zenodo. For the second option please also have a look at the Zenodo notebook.
[25]:
ref_path = 'ref_model/'
vae.save(ref_path, overwrite=True)
reference_latent.write(ref_path + 'ref_latent.h5ad')
Construct hierarchy for the reference using scHPL
First, we concatenate all cell type labels with the study labels. This way, we ensure that the cell types of the different studies are seen as unique.
Warning: Always ensure that the cell type labels of each dataset are unique!
[26]:
reference_latent.obs['celltype_batch'] = np.char.add(np.char.add(np.array(reference_latent.obs['cell_type'], dtype= str), '-'),
np.array(reference_latent.obs['study'], dtype=str))
Now, we are ready to learn the cell-type hierarchy. In this example we use the classifier='knn', this can be changed to either a linear SVM ('svm') or a one-class SVM ('svm_occ'). We recommend to use the kNN classifier when the dimensionality is low since the cell-types are not linearly separable anymore.
The option dynamic_neighbors=True implies that the number of neighbors changes depending on the number of cells in the dataset. If a cell-type is small, the number of neighbors used will also be lower. The number of neighbors can also be set manually using n_neighbors.
During each step of scHPL, a classifier is trained on the datasets we want to match and the labels are cross-predicted. If you’re interested in the confusion matrices used for the matching, set print_conf=True. The confusion matrices are also saved to .csv files then.
For more details about other parameters, take a look at the scHPL GitHub
[27]:
tree_ref, mp_ref = sca.classifiers.scHPL.learn_tree(data = reference_latent,
batch_key = 'study',
batch_order = ['Freytag', 'Oetjen', 'Sun'],
cell_type_key='celltype_batch',
classifier = 'knn', dynamic_neighbors=True,
dimred = False, print_conf= False)
Starting tree:

Adding dataset Oetjen to the tree
Updated tree:

Adding dataset Sun to the tree
Updated tree:

Use pretrained reference model and apply surgery with a new query dataset to get a bigger reference atlas
Since the model requires the datasets to have the same genes we also filter the query dataset to have the same genes as the reference dataset.
[28]:
target_adata = target_adata[:, source_adata.var_names]
target_adata
[28]:
View of AnnData object with n_obs × n_vars = 10727 × 2000
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue'
layers: 'counts'
We then can apply the model surgery with the new query dataset:
[29]:
target_adata = target_adata.copy()
[30]:
model = sca.models.SCVI.load_query_data(
target_adata,
ref_path,
freeze_dropout = True,
)
INFO File ref_model/model.pt already downloaded
[31]:
model.train(max_epochs=50)
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Epoch 50/50: 100%|██████████████████| 50/50 [00:26<00:00, 1.89it/s, loss=975, v_num=1]
[32]:
query_latent = sc.AnnData(model.get_latent_representation())
query_latent.obs['cell_type'] = target_adata.obs["final_annotation"].tolist()
query_latent.obs['batch'] = target_adata.obs["batch"].tolist()
And again we can save or upload the retrained model for later use or additional extensions.
[33]:
surgery_path = 'surgery_model'
model.save(surgery_path, overwrite=True)
query_latent.write('query_latent.h5ad')
Get latent representation of reference + query dataset and compute UMAP
[34]:
target_adata.obs.study = "10X"
[35]:
target_adata
[35]:
AnnData object with n_obs × n_vars = 10727 × 2000
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue', '_scvi_batch', '_scvi_labels'
uns: '_scvi_uuid', '_scvi_manager_uuid'
layers: 'counts'
[36]:
adata_full = source_adata.concatenate(target_adata, batch_key="ref_query")
adata_full
[36]:
AnnData object with n_obs × n_vars = 32484 × 2000
obs: 'batch', 'chemistry', 'data_type', 'dpt_pseudotime', 'final_annotation', 'mt_frac', 'n_counts', 'n_genes', 'sample_ID', 'size_factors', 'species', 'study', 'tissue', '_scvi_batch', '_scvi_labels', 'ref_query'
var: 'highly_variable-0', 'means-0', 'dispersions-0', 'dispersions_norm-0', 'highly_variable_nbatches-0', 'highly_variable_intersection-0'
layers: 'counts'
[37]:
full_latent = sc.AnnData(model.get_latent_representation(adata=adata_full))
full_latent.obs['cell_type'] = adata_full.obs["final_annotation"].tolist()
full_latent.obs['batch'] = adata_full.obs["batch"].tolist()
full_latent.obs['study'] = adata_full.obs["study"].tolist()
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
[38]:
sc.pp.neighbors(full_latent)
sc.tl.leiden(full_latent)
sc.tl.umap(full_latent)
[39]:
full_latent.obs['study'] = full_latent.obs['study'].astype('category')
full_latent.obs['study'].cat.add_categories(['0'], inplace=True)
full_latent.obs['study'].cat.reorder_categories(['Oetjen', 'Sun', 'Freytag', '0', '10X'], inplace=True)
sc.pl.umap(full_latent,
color=['study'],
frameon=False,
wspace=0.6, s=25,
palette=sns.color_palette('colorblind', as_cmap=True)[:5],
save='study_query.pdf'
)
WARNING: saving figure to file figures/umapstudy_query.pdf
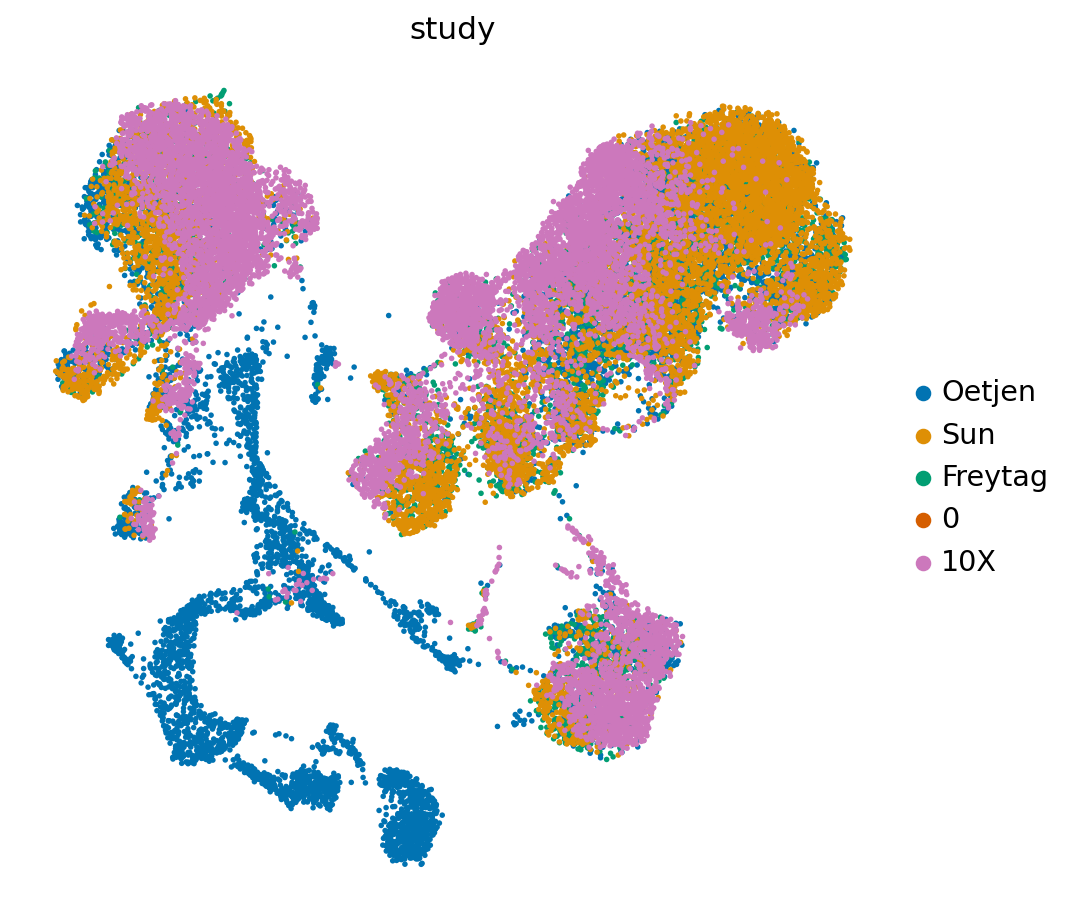
[40]:
full_latent.obs['ct_low'] = 0
idx = ((full_latent.obs.cell_type == 'CD4+ T cells') |
(full_latent.obs.cell_type == 'CD8+ T cells'))
full_latent.obs['ct_low'][idx] = 'T cells'
idx = ((full_latent.obs.cell_type == 'CD10+ B cells') |
(full_latent.obs.cell_type == 'CD20+ B cells'))
full_latent.obs['ct_low'][idx] = 'B cells'
idx = ((full_latent.obs.cell_type == 'CD14+ Monocytes') |
(full_latent.obs.cell_type == 'CD16+ Monocytes') |
(full_latent.obs.cell_type == 'Monocyte progenitors'))
full_latent.obs['ct_low'][idx] = 'Monocytes'
idx = ((full_latent.obs.cell_type == 'Erythrocytes') |
(full_latent.obs.cell_type == 'Erythroid progenitors'))
full_latent.obs['ct_low'][idx] = 'Erythrocytes'
idx = ((full_latent.obs.cell_type == 'Monocyte-derived dendritic cells') |
(full_latent.obs.cell_type == 'Plasmacytoid dendritic cells'))
full_latent.obs['ct_low'][idx] = 'Dendritic cells'
idx = full_latent.obs.cell_type == 'HSPCs'
full_latent.obs['ct_low'][idx] = 'HSPCs'
idx = full_latent.obs.cell_type == 'Megakaryocyte progenitors'
full_latent.obs['ct_low'][idx] = 'Megakaryocyte progenitors'
idx = full_latent.obs.cell_type == 'NK cells'
full_latent.obs['ct_low'][idx] = 'NK cells'
idx = full_latent.obs.cell_type == 'NKT cells'
full_latent.obs['ct_low'][idx] = 'NKT cells'
idx = full_latent.obs.cell_type == 'Plasma cells'
full_latent.obs['ct_low'][idx] = 'Plasma cells'
/tmp/ipykernel_1063462/1546695568.py:5: SettingWithCopyWarning:
A value is trying to be set on a copy of a slice from a DataFrame
See the caveats in the documentation: https://pandas.pydata.org/pandas-docs/stable/user_guide/indexing.html#returning-a-view-versus-a-copy
full_latent.obs['ct_low'][idx] = 'T cells'
[41]:
sc.pl.umap(full_latent,
color=['ct_low'],
frameon=False,
wspace=0.6, s=60,
palette=sns.color_palette('colorblind', as_cmap=True),
save='cp_query.pdf'
)
WARNING: saving figure to file figures/umapcp_query.pdf

[42]:
for s in np.unique(full_latent.obs.study):
ref_s = cp.deepcopy(full_latent)
ref_s.obs.ct_low[full_latent.obs.study != s] = np.nan
sc.pl.umap(ref_s,
color=['ct_low'],
frameon=False,
wspace=0.6, s=60,
palette=sns.color_palette('colorblind', as_cmap=True), title=s,
save=s+'_query.pdf'
)
WARNING: saving figure to file figures/umap10X_query.pdf

WARNING: saving figure to file figures/umapFreytag_query.pdf

WARNING: saving figure to file figures/umapOetjen_query.pdf

WARNING: saving figure to file figures/umapSun_query.pdf

Updating the hierarchy using scHPL
If the cells in the query dataset are labeled, we can update the hierarchy using scHPL. If the cells are unlabeled, we can predict their label (see section below).
Again, we first have to ensure that the labels of the cell-types are unique
[43]:
full_latent.obs['celltype_batch'] = np.char.add(np.char.add(np.array(full_latent.obs['cell_type'], dtype= str), '-'),
np.array(full_latent.obs['study'], dtype=str))
Now, we are ready to update the cell-type hierarchy. It is important to use the same classifier settings here as used before. Furthermore, it is important to indicate which batches are already in the tree (batch_added) and which you want to add to the tree (batch_order).
[44]:
# First make a deep copy of the original classifier to ensure we do not overwrite it
tree_rq = cp.deepcopy(tree_ref)
tree_rq, mp_rq = sca.classifiers.scHPL.learn_tree(data = full_latent, batch_key = 'study',
batch_order = ['10X'],
batch_added = ['Oetjen', 'Freytag', 'Sun'],
cell_type_key='celltype_batch',
tree = tree_rq, retrain = False,
classifier = 'knn',
dimred = False)
Starting tree:

Adding dataset 10X to the tree
Updated tree:

Predicting cell-type labels using scHPL
If the cells in the query dataset are unlabeled or if you’re interested in comparing the transferred labels to your own annotations without updating the hierarchy, you can predict the labels with scHPL.
[45]:
query_pred = sca.classifiers.scHPL.predict_labels(query_latent.X, tree=tree_ref)
Using the evaluate.heatmap() function, the predictions can be compared to other annotations
[46]:
sca.classifiers.scHPL.evaluate.heatmap(query_latent.obs['cell_type'], query_pred, shape=[8,5])
